By Sam DeLuca, Ph.D. With Steven Lewis, Ph.D. and Lucas Nivon, Ph.D.
June 2023
In our previous article in March 2023, we discussed our experience benchmarking publicly available AI protein design tools and exploring their real world strengths and weaknesses. Here, we want to be more speculative and lay out a vision for how we think these AI methods will impact the future development of protein engineering and the resulting clinically meaningful biologics.
AI Structure Prediction as a rapid hypothesis test
While AlphaFold2, OpenFold, and ProteinMPNN have led headlines with their remarkable effectiveness at making accurate protein structure and sequence predictions, an underappreciated impact of these tools is their speed. These tools have reduced the amount of computational time required for comparative modeling (protein structure prediction via traditional homology and physics methods) and protein design tasks. As a rough example, a comparative modeling job which would have taken thousands of hours of CPU time using Rosetta in 2010 and hundreds of hours of CPU time in 2018 now takes only 1-2 hours of CPU/GPU time using AlphaFold2 or OpenFold with a reasonably powerful GPU like a Tesla T4. The most obvious impact of this drastic reduction of compute time is that researchers can now perform modeling experiments on a vast scale, such as the AlphaFold Protein Structure Database.
A somewhat less obvious impact of the reduction of compute time is the reduction in level of effort, cost, and impact of homology modeling of a single protein. Although academic use of homology modeling via free online servers like Robetta has been straightforward for 20 years, license and data security requirements have kept the tool out of the hands of industry. More recently, Cyrus Bench alleviated this. Still, running homology modeling yourself on local secure resources was a major effort, requiring scientists with specialized training and substantial analysis and post-processing effort. This level of effort limited the use of comparative modeling in industrial settings, where access to large scale compute hardware is more expensive relative to academic research facilities.
Current ML tools have effectively eliminated this limitation. When comparative modeling is trivial both from a technical and a cost standpoint, it becomes an obvious first step in any molecular modeling project. AlphaFold2 and OpenFold do not always produce useful output, and their output needs to be carefully scrutinized before use, but for native proteins the vast majority of the time they result in a model which can provide at least some useful structural insights, and frequently produce models that are directly useful in protein engineering. The ability to produce usable models cheaply and rapidly also opens the door to novel uses of comparative modeling, such as validation of de novo protein designs, as we have seen now extensively from the Baker Lab at the University of Washington.
We still need physics
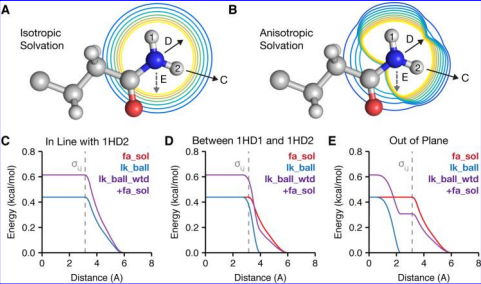
The Lazaridis-Karplus solvation model used by Rosetta (Alford et al)
Despite the power of molecular modeling techniques for rapidly predicting sequences and structures, they remain inferior to statistical physics based methods at ranking models by stability or activity and differentiating between models of very similar sequence. For this, it is still important to use a more traditional physics or knowledge based scoring function (such as the Rosetta hybrid statistical/physical energy function or a “pure physics” molecular mechanics force field, or a combination thereof).
Additionally, machine learning methods are generally incapable of modeling outside of the domain of their training data, and thus for the foreseeable future it will be necessary to use more traditional methods to model novel small molecule and Non-Canonical Amino Acid (NCAA) chemistry. Small molecule and NCAA modeling are therefore of high priority for the OpenFold Consortium which Cyrus co-founded with Genentech/Prescient, Outpace, and Arzeda.
We still need experimental validation in the biochemistry lab and in animals
GFP tagged proteins expressed in the Cyrus lab
A designed protein operates in a complex biological environment, and in most medically relevant cases that environment is only partially understood. For this reason, experimental validation of designs will always be a necessary component of ML driven protein design, regardless of the accuracy of the ML model, because those ML models are trained on examples of purified proteins in artificial buffers, not on the behavior of proteins inside a cell, let alone a protein inside a living mouse or human.
In Cyrus’ experience, to make the best use of computational protein design tools it is necessary to have an efficient, closely collaborative loop between computational design and experimental validation. If well executed, this loop creates a virtuous cycle in which the results of the experimental validation provide data and insights which are used to fine-tune future rounds of computational design, and the computational design provides insights into the structure and chemistry of the designed proteins that can improve experimental methods. Along the way, the core algorithms are expanded and become more capable for future projects.
Establishing the “primitives” of protein engineering
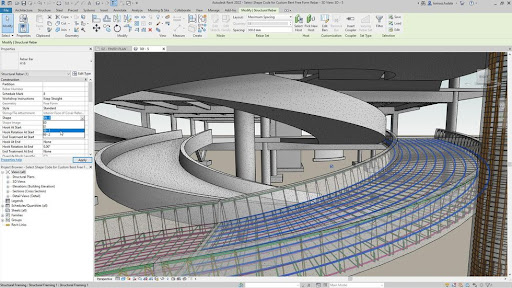
Given these new tools, it’s worth taking a step back and considering what a comprehensive tool for protein engineering might look like in the near future. To begin with, let us compare the basic user interface of Cyrus Bench, the GUI protein engineering tool which Cyrus developed before any of the current ML tools existed, and Autodesk, an industry standard CAD tool:
The basic UI concept between these two applications is intentionally similar, with a model (a building for Autodesk, a protein structure for Bench) being designed at the center of the user interface, and a palette of tools to manipulate that structure (e.g. extrusion of a 3D shape for Autodesk, loop rebuilding for a protein in Bench.
Inspection of the actual tools available, however, indicates a major difference between the user interactivity in the two tools – Autodesk is fast and responsive, Bench is mostly running slow calculations on its huge cloud back end. The tools available in Autodesk (and in most other mature engineering, art, and design tools) are relatively simple primitives like “extrude”, “rotate”, etc. The user is given a palette of fast running primitives to manipulate their model, and a larger set of analysis and measurement tools to analyze and validate that model. The vast majority of the tools in Bench take minutes to hours to run, even with tens to hundreds of CPUs calculating on the backend, and therefore most user interaction is asynchronous.
(Note: The authors of this blog post is a structural biologist and software engineer, not a mechanical engineer and has only a superficial understanding of actual CAD software).
The advantage of simpler individual tools is that they allow the user more flexibility and interactive reasoning in their usage of the tool. From a user experience perspective, these tools need to run very quickly in order for them to be usable. Historically, molecular modeling methods have been far too long-running and computationally expensive for this sort of simple tool user interface to be feasible. However, the dramatic reduction in the amount of time necessary to perform a single modeling action over the past several years opens the possibility of making far more interactive and fluid protein modeling tools than were previously possible.
In a hypothetical purely AI-driven future for protein design, then,fasst-running tools could be exposed to domain-expert users directly. Assuming those tools have been vetted for useful outputs, as we discussed in our last blog post, those users will be able to rapidly iterate on a protein design while having a reasonable expectation that the outputs will behave as desired in the lab.
You get what you train for: Data will drive progress
As of this writing in mid 2023, some of the tools for protein design remain in the domain of physics/statistics, while some are now fully AI/deep learning tools that have replaced older tools. Roughly speaking this process is 50% complete. Structure prediction and design of full de novo proteins and heavily-redesigned proteins (changing more than 50% of the residues in a protein) are now all tools in the realm of AI. Redesign of natural proteins is still mostly not AI driven, especially if a crystal structure exists of the starting protein – this includes the design for point mutations to increase stability or affinity, design of solubility, or design of glycosylation.
Over time as datasets grow, and new ways of collecting large-scale data are developed, we expect the applicable domain of AI methods in molecular modeling to grow. This seems to be happening with protein stability. Work such as Gabe Rocklin’s at the University of Chicago in “mega-scale protein design” is paving the way to improved AI driven protein stability models. We see this area as a very fruitful collaborative area between academia and industry, where open data sources can lead to massively improved open models, including through the AI consortium we cofounded, OpenFold. Other datasets in protein/ligand binding or antibody structure could also lead to new models for small molecule drug development and biologics development, respectively.
All of these AI models are driven by the large scale, accurate, physiologically-relevant data that underlie them. David Baker and Minkyung Baek theorized about some of these future directions in Nature in early 2022. In areas where we can collect millions of high-quality, diverse data points, we expect AI models to eventually replace older models. In other areas where it may be difficult or impossible to generate such data, we may continue to use physical models, hybrid statistical/physical models, or even quantum mechanical models.
The generation of the underlying datasets, whether across large sets of proteins or just for one family of proteins, will drive further general advances and help optimize specific proteins as drugs. Cyrus’ principle focus for dataset generation is deep mutational scanning for individual proteins of therapeutic interest, so we can create superior biologics that are optimized across sequence space for maximal efficacy, high specificity, and few liabilities that could hinder clinical development.
Going to the clinic
The measure of our field’s work in AI-driven biologics discovery will be in it’s clinical impact, not in the diversity of our models or our ability to generate data. Here we’ve outlined the huge changes that AI structure prediction and de novo design have already made, the continuing role of statistics and physics in biologics modeling, the necessity of iterations between algorithm and laboratory, the changes resulting from faster AI methods, and where our core AI models will be developing next. We are excited to move superior biologics into the clinic at Cyrus, and for the progress that the next 5 years will bring across the BioPharma industry as a variety of firms bring new AI designed drugs to clinical trials and into patient’s lives.
https://cyrusbio.com/wp-content/uploads/CyrusLogoStaggeredWhite.svg00Thomas Tuonghttps://cyrusbio.com/wp-content/uploads/CyrusLogoStaggeredWhite.svgThomas Tuong2023-06-05 11:40:372023-06-07 11:15:49The future of AI in biologics drug discovery
Protein AI tools are useful for aggressive protein design, while physics based tools continue to be required for re-design of natural proteins for most cases
By Steven Lewis, Dan Farrell, Brandon Frenz, Ragul Gowthaman, Ryan Pavlovicz, David Thieker, Indigo King, Sam DeLuca, Yifan Song, and Lucas Nivon
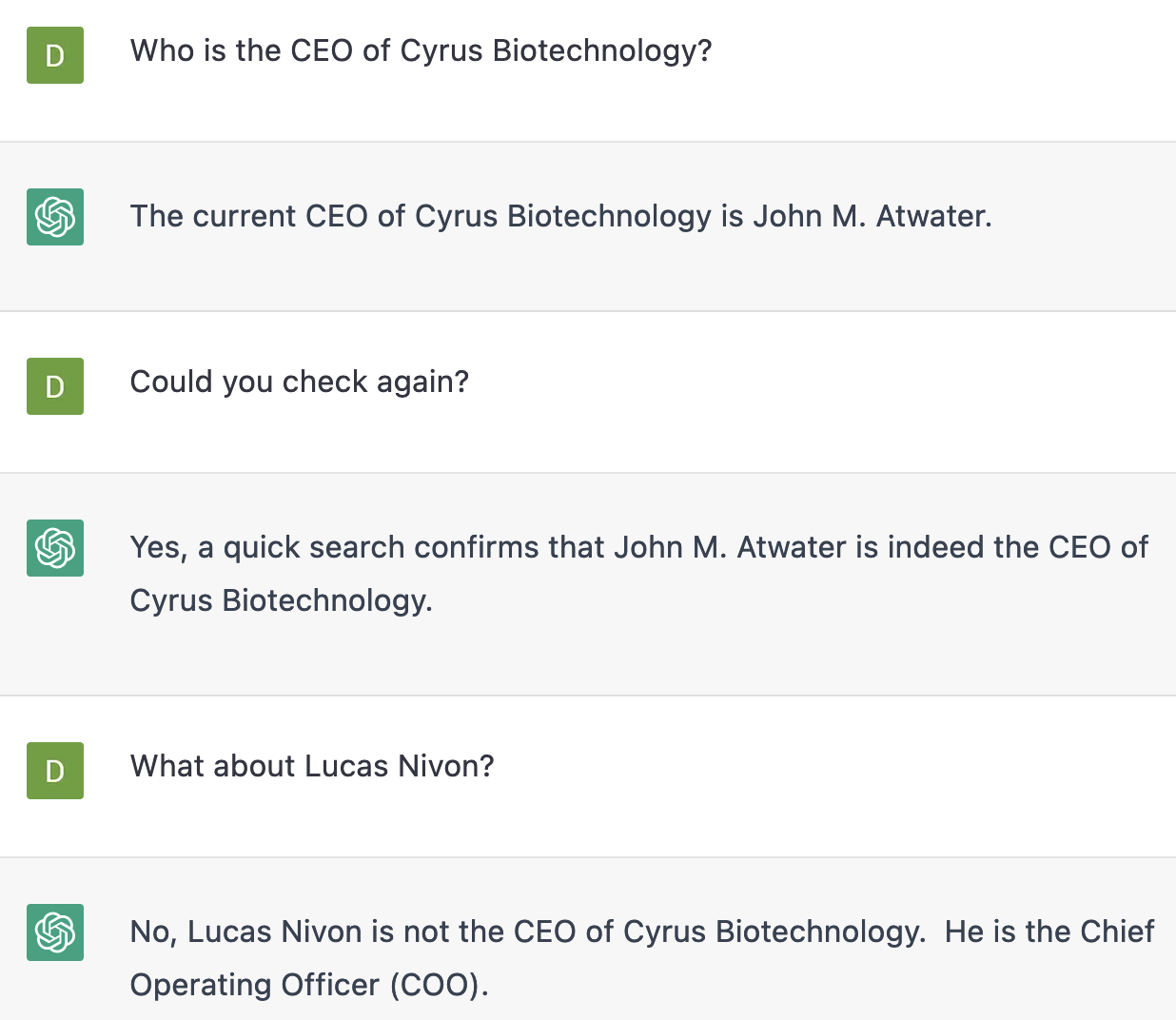
As new tools come online daily applying deep learning architectures to new problems, the field of artificial intelligence is replete with claims, from the fairly pedestrian to aggressive claims of having “solved” entire fields of human endeavor. Some of this hype is the result of academic creators’ excitement about their progeny, some is commercial marketing…and some is of course highly valid and well supported by the data! Taking ChatGPT as an example, use of the tool will lead the savvy user to the conclusion that while it is excellent at producing well structured English text, the tool’s grasp on reality is much weaker – stories of grammatically correct but factually nonsensical interactions abound, for example in health and writing plagiarism. The media and PR rhetoric for “this tool writes English” is very valid, but it’s an open question whether one should trust it to write something that is factually correct, true, and well supported by other publications and data.
ChatGPT is wrong – Lucas Nivon is the CEO, Cyrus has never had a COO, and as far as we are aware John Atwater is a fictitious person in this context. (ChatGPT is an evolving tool and your responses may vary if you try this experiment yourself.)
Written English is a great area to evaluate AI fallibility because even a layperson can easily judge for quality – but these problems presumably recur within other domains of AI tooling, including the powerful tools for protein folding and sequence design like AlphaFold2, OpenFold, and ProteinMPNN. Repeated experiments and benchmarking have shown they are good at their most direct intended use cases. In the case of AlphaFold2/OpenFold, that is prediction of the structures of naturally occurring sequences, using “blinded” 3D protein structures released as part of the CAMEO project to test the algorithms on input sequences that have never been seen before. For ProteinMPNN, that is de novo or nearly de novo design problems – the creation of new protein sequences, followed by validation to determine whether those proteins have the intended structure. Most scientists agree that these tools are great for some use cases, but it is necessary to ask where things might go wrong, to understand when these tools produce a beautifully written essay, and when they produce the protein equivalent of our fictitious “John M. Atwater”. AlphaFold2/OpenFold will simply take “a protein sequence” as input – is it wise to query these with any sequence you might have, or are there some wrong “questions” to ask? For ProteinMPNN, are all design problems equally valid, or only certain types of proteins designable, or only large sets of mutations computable?
One way to interpret the collective decades of computational biophysics at Cyrus is that we have an enormous amount of experience in knowing when a computer is lying to us in the form of a nonsensical, non-physical, or otherwise unrealistic protein structure. With Rosetta specifically, we have world class experience in understanding when to take out the computational in “computational protein design”. For example, for many years Rosetta had a tendency to place too many Trp residues on the surface of proteins, or to make mistakes with the backbone angles of certain aromatic residues – the latter set of failures even had a specific set of flags designed by Nobu Koga to minimize these failures that only a handful of human experts could spot by eye. We’ve been spending time expanding this type of “protein design quality control” to the realm of AI tooling to understand the failure modes and limitations of popular tools – to know where we can trust the AI output as is (like a reasonable rhyming poem written by ChatGPT), when we should be dubious or filter the output (like the plagiarism problem), and when we should simply not trust the AI output whatsoever (“John M. Atwater is the CEO of Cyrus”).
AI structure prediction
Before AlphaFold1 was released in 2018, Rosetta was reliably a strong bet for protein structure prediction in all contexts, especially homology modeling problems via Hybridize (protein structure prediction where relatively high sequence identity homologs exist in the pdb, over 30% sequence identity). For folding of soluble native monomers, classic Rosetta algorithms and their competitors lost their crown to AlphaFold1; the crown is currently shared between AlphaFold2 and OpenFold. These are sibling algorithms independently trained with similar performance; the chief difference is that OpenFold has been tuned to run faster on commercially available chips from NVIDIA, and its training code is released in the public domain allowing anyone to fine-tune the algorithm for their protein problems of interest. Because these algorithms are siblings, and perform similarly in experiments mentioned here, we will abbreviate the pair AF2/OF.
These tools were trained on the wealth of structurally determined proteins in the PDB, most of which are wild type (not mutated from the natural sequence cloned out of an organism) and all of which were sufficiently well behaved to be expressed and isolated in quantity for structure determination. Any bench scientist will tell you – not all proteins are so neatly behaved, so these proteins are by construction a biased sample of the actual proteome or structureome, where poorly behaved proteins, or even proteins that are only well structured in their physiological context, are simply not included.
In the context of native proteins, AlphaFold2 already does valuable if unexpected things for predicting inherently disordered regions – it leaves those regions as “spaghetti” looping all over the model, to use the technical term. These types of structures are extremely rare in the PDB, as the disorder is not compatible with the ordered nature of a crystal. This type of loop-heavy output turns out to be rather a bonus, in that AlphaFold2 is useful as a disordered structure predictor (for example, here’s an early review). However, the output for “this probably doesn’t have structure” type of protein sequence being so wild raises the question of “how SHOULD the tool signal that the input sequence does not produce well ordered structure”? To the untrained eye, these looped outputs might seem sensible, but to Cyrus scientists it is immediately apparent when a disordered region is being predicted, even though AF2 does not alert the user to this. It is also obvious to the biophysicist, but not necessarily the layperson, that the spaghetti regions are useful as space-filling envelopes but that the coordinates themselves are invalid. The lesson here is first, that the AI tool gives reliably incorrect results (the atomic coordinates are wrong), and second, that the results can be useful anyway in a different, less obvious context.
Protein structure prediction: Small changes to natural proteins
The next step past disordered structure prediction, and one with which Cyrus is deeply concerned, is how these tools ought to respond to narrowly non-native sequence inputs. Small mutations to natural proteins that are intended to improve the efficacy or safety of a protein biologic candidate are the bread and butter of much of the $300B market for biologics, and at the core of Cyrus’s drug discovery efforts.
In Cyrus’s view, after having worked with over 100 BioPharma firms including 13 of the top 20 BioPharma firms by annual sales, this type of optimization of natural proteins by introducing small numbers of mutations or creating novel fusion proteins with other natural proteins represents 99% of the therapeutic use of protein design tools in 2023, and therefore is where the most near-term impact on patient health will be delivered by AI tools.
The future will bring huge growth in that 1% of de novo designed proteins that are unrelated to natural proteins, and Cyrus’s view is that in twenty years a large majority of biologics in clinical trials are likely to be de novo designed optimal proteins, but for patients looking for cures in the next few years, the modification of natural proteins is of paramount importance. Nature Biotechnology recently covered this broader de novo space in a news article, including an interview with Cyrus’s CEO, Lucas Nivon. Next-next-generation therapeutics on de novo protein tech is coming, but the next generation will be more nativelike, which forces us to ask how well AI tooling works on nearly native proteins.
The difference between well folded protein and misfolded, aggregated crud is often a single mutation. How do AlphaFold2 and OpenFold respond when challenged with nearly-native proteins known to be poorly behaved in solution?
The answer turns out to be “not well”. Cyrus has a deep mutational scan data set for a certain human signaling protein, from which we selected deleterious mutations – because this is the subject of Cyrus IP, we’ll call this protein FOO. An ideal response from a structure prediction tool, when presented with a sequence that does not form a stable structure, would be either a non-structure output (essentially an error message – “this does not fold”), or at worst an output that is visibly malformed relative to the parent sequence’s structure, so a human user could immediately recognize “something has gone wrong, this is very likely a bad mutation”. A usable, but not ideal, response would be a glaring change to the confidence statistics, even if the predicted fold were maintained.
The actual response from AlphaFold2 and OpenFold to known destabilizing mutations is quite tepid. If you load 50 proven deleterious mutations simultaneously onto the <150 residue FOO sequence, AlphaFold2 and OpenFold show only mild deficits in pLDDT and some extra noise and variability in the predicted model population, even though this is an extremely poor protein that has no hope of expressing in vitro. When presented with a single deleterious mutation at a time there is basically no response, even though we know that each of those results in a protein that will not express in vitro. We interpret this insensitivity to deleterious mutations as template bias: WT FOO protein structure would have been present in the training data sets, and of course is present in the MSAs used for structure prediction. The structure prediction algorithm appears to place all its faith in the MSA and ignores the sequence deviations introduced by our known deleterious mutations. This makes sense for predicting native proteins known to be well-behaved enough to be useful to a cell, but unfortunately means these tools are not useful for judging the structural impact of mutations.
In theory this behavior makes sense, given the training corpus. AF2/OF are trained on the pdb structures which, as we mentioned above, are a pre-selected set of proteins that are well behaved enough to have their structures solved in the first place. AF2/OF are not structurally trained on the “bad” proteins that did not express, or expressed poorly, or did not crystallize because they are too dynamic or disordered. Their MSA inputs and related training do not “know” much about protein sequences that are not functional in nature. The models do not “know” anything about badly behaved proteins, and therefore they cannot label proteins as poorly behaved. This suggests a path to improving this behavior by including these “bad” protein sequences in training data.
This image represents simulated data showing approximately the number and distribution of deleterious mutations (spheres) to which AF2/OF did not respond, either in combination or individually.
How can we rescue the performance of these tools for predicting the behavior of deleterious mutations?
The quality and quantity of the MSA data given to an AI folding tool is often directly correlated with the tool’s confidence and accuracy. In this context, we can reference our previous example and ask: why do these tools fail to recognize the structural changes that should be induced with deleterious mutations? Our findings suggest that this characteristic is partially due to how tolerant MSA constructing tools are to mismatches and partially due to the lack of structural data derived from peptides that may or may not express, or be functional.
To reassess AF2’s ability to predict changes induced by deleterious mutations, in the following figure we asked AF2 about the BAR enzyme (again, this is internal Cyrus IP, so we will anonymize for the purposes of this blog post), with and without 2 proline mutations in the middle of 2 helices. We found that the unmodified AlphaFold2 tool happily predicted (with relatively high confidence!) that a double proline mutation would have no effect on the local helical structure, and also found that the MSAs created in each of the two AlphaFold2 experiments were nearly identical. If you think about this experiment with the MSA as the input in mind, we have tasked this tool to predict two wildly different results, given inputs that are >99% similar. Although all your biophysics alarm bells should be going off at full volume after seeing such a result – two Pro mutations will break almost any local structure – we should not only blame AlphaFold2 for this result. AlphaFold2 was misled into thinking this was ‘just another protein family MSA’ by the MSA tool that ignored this mismatch in order to retain its gapless alignment. Since we can’t force AlphaFold2 to read Leninger’s Principles of Biochemistry, to give it a fighting chance at predicting deleterious mutations we can mask out regions of the MSA to coerce the tool to create predictions based on its internal biophysical intuition alone.
To test this we asked AF2 to again fold the BAR enzyme with and without 2 proline mutations in the middle of 2 helices, but this time we masked a small region of the MSA near the position of these mutations. We find that in this environment AF2 is able to produce an accurate model for the native sequence, but has diminished confidence. When AF2 predicts the double proline mutant however, it completely blows apart the helix and instead predicts an unstructured loop – the result we would expect, short of totally unfolding the protein. In other words, by masking out the MSA we find that AF2/OF will now predict a structural shift as a result of these highly damaging Pro mutations, which now fits with experimental reality. It is clear that both cases with masked MSAs lead to lower confidence predictions – which suggests that in the tool’s structure prediction confidence may be too much a function of MSA coverage/quality and too little a function of physical plausibility.
Protein language models and protein structure prediction
Meta’s (Facebook) ESMFold tool is also a major player in the AI structure prediction field, with the advantage that it does not require an MSA input, but the disadvantage that its outputs are on average less accurate than MSA-input outputs from AF2/OF. ESMFold uses a different first step, with a language model replacing the MSA step that AF2/OF use. The prediction step is using OpenFold with embeddings from the language model.
We asked two questions about ESMFold. First, given its lack of MSA dependence, does it do any better at the deleterious point mutations experiment? Unfortunately the answer is no, although we are less sure why. Second, given that it is faster than AlphaFold2 or a full OpenFold run (because the language model step is faster than generating MSAs), are the results of lower quality? In our hands, ESMFold generally produces somewhat lower quality results, as measured by RMSD to known structures – but it also tracks neatly with lower confidence. ESMFold is a tool that is faster, correct (under 2 Ångstroms RMSD) less often, but also knows when it is incorrect (on native sequences) – suggesting that it is a fine first pass tool if you want to look at many native proteins at once.
Plotted above is pLDDT vs. RMSD for a subset of 67 sequences/structures from CAMEO that are modeled using all three methods. AF2 and OF tend to produce higher pLDDT “confidence” scores even if the structure is rather incorrect (over 5 Ångstroms RMSD). In contrast, ESMFold produces slightly more incorrect structures but those structures have lower pLDDT/“low confidence”, so the model is correctly flagging when the answer is likely wrong.
Protein modifications, glycosylation, and other chemistries
A final limitation to consider with AI structure prediction tools, especially for real-world drug development where post-translational modifications such as PEG or glycosylation are added in order to produce a clinically useful drug, is the narrowness of their chemical universe. The tools discussed here are unable to deal with modified or noncanonical amino acids, small molecule ligands, nucleic acids, or carbohydrates (glycans in the protein context). Physics-based algorithms like Rosetta’s Hybridize for homology modeling are no longer cutting edge for simple native proteins, but they are still the best game in town for more complex problems involving more diverse biomolecules – for example, Cyrus has an automated integrated tool using OpenEye software for small molecule physical parameters combined with Rosetta Hybridize to produce high-quality models of protein/ligand complexes, we call this “Ligand HM”. We are confident that over time the AI tools available in the open source or publicly disclosed world will fill in some of these gaps either with AI or with hybrid AI/physics solutions. Baker and Baek described some of these gaps in a short Comment in Nature Methods in 2022. Multiple academic groups are working on a variety of these problems as we speak.
By the same token, classical mutational DDG (free energy (delta-G) of a point mutation/delta, hence delta-delta-G) calculations are still better at evaluating the effect of single or small numbers of mutations than the AI tools. The AI folding tools tend to get lost in the big picture and ignore the details of a mutation.
AI and protein design at the large and small scale
ProteinMPNN has demonstrated extremely impressive results for de novo (no relation to natural proteins) and nearly de novo (some relation to natural proteins) design problems. The commercial application of protein design is currently more grounded in the biochemistry of natural proteins: usually you already have a protein that is nearly right and you want to modify it minimally to improve some specific behavior, like making a minimal number of mutations to increase stability, binding affinity to the disease target, or increase serum half-life. Many problems are easy to cast in terms of mutational DDG on an existing protein, but much more difficult to cast as a fully de novo sequence in novel scaffolds.
We were curious if the ProteinMPNN “scorefunction” (its confidence) tracked with the same deep mutational scan data as above – can MPNN predict which individual mutations are beneficial and which are harmful? The correlation goes in the right direction, but it is not impressive in comparison with pre-existing Rosetta/physics based techniques. This experiment is hedged with caveats – for example the native backbone is probably not accurate for deleterious mutations, so a simple backbone-threaded input is maybe set up to perform poorly in ProteinMPNN. ProteinMPNN is not a tool built for judging point mutations, and indeed it does only a mediocre job at that task when applied directly without modification, as we’ve done here.
The correlation between deep mutational scan scores for protein FOO and MPNN scores for the same sequences is unimpressive, although with some trend in the correct direction.
We’ve also aimed to test ProteinMPNN on tasks more aggressive than single point mutations but less aggressive than full protein design – for example redesign of a protein core while leaving the surface intact. The results concerned us in several ways. In this example, we were designing 41 out of 173 residues in the BAZ helical bundle. ProteinMPNN was trained with an atom-positional fuzzing in effect, which should make it insensitive to small changes in input backbones. We were nevertheless running this experiment in quintuplicate, with 5 AlphaFold2-generated backbones (BAZ has a crystal structure but the loops are missing, and AlphaFold2 is a cheap way to fix that particular problem) regularized through Rosetta Relax.
We noted a pair of related problems: first, that ProteinMPNN happily packed 4 glutamate residues facing each other in the core of the protein (again, your biophysics alarm bells ought to be screaming); and second, that it only liked to do this in 2 of the 5 input models. It is plausible that ProteinMPNN thinks some sort of metal binding site ought to go here, which would be the only explanation for charges like this in a protein core. Given that ProteinMPNN does not know what metal ions are, nor can it place them in its sequence output, it’s still worrying. Even if a metal site could go there, a protein design experiment usually has that as an explicit goal, not a random happenstance. Our best hypothesis for the backbone dependence of this effect is that ProteinMPNN is simply not performing well on partial redesigns where “partial” is not the vast majority of the protein. ProteinMPNN’s published native sequence recovery results prove it is perfectly happy to keep native sequence where appropriate, but we assert that the tool only performs well when that is a choice ProteinMPNN is making, rather than one the experimenter forces by removing design positions.
Above, note the unrealistic quadruple E mutation in the protein core. Below, note that it is only common in 2 of the 5 input backbones, even though all are mutually low RMSD to each other and the native BAZ and geometrically normal.
From these experiences we conclude that ProteinMPNN should be used only when it can alter nearly all of the protein sequence – in other words if one is allowing some large fraction (we are guessing 80%) of a protein to vary, MPNN should perform well. In the authors’ hands, in our hands, and in many experiments from other researchers it does a great job when nearly an entire protein is open for mutation, even when it chooses to maintain native sequence. We find if you aren’t letting it “think about” mutations at most positions it probably won’t work well, and consequently if your intended use can’t tolerate the possibility of mutations scattered throughout the protein it may not be the right tool to use. For project confidentiality reasons we can’t provide detailed data on this point (we can’t even provide a fake name as we ran out of metasyntactic variables), but we find similar results for cases of nearly full protein redesign to what the authors found. Cases where >80% of the protein is mutable still come back with designs where ~50% of the sequence is native, and most of the expressed proteins are both soluble and active.
Protein design for drug discovery using AI protein design tools
At Cyrus we are incorporating the AF2/OF tools and ProteinMPNN tools into our workflows, alongside our own Rosetta tools and combined AI/Rosetta software we have written for other protein tasks. For example, we are using AF2/OF to validate novel homologs identified by sequence bioinformatics searches for protein targets of interest prior to screening a small number in the lab for enzyme or binding activity. We no longer use ProteinMPNN for small redesign problems, but are using it for exploratory design when over 80% of the residues in a protein sequence can be left free.
At Cyrus we design for immunogenicity reduction, solubility, lack of aggregation, protein stability, serum stability, serum half life, binding affinity (to a target protein or substrate), and binding specificity (to a target protein or substrate). For most of those we are using a combination of Rosetta and specialized Rosetta/AI combination tools to introduce a relatively small number of mutations, thereby keeping close to a natural sequence and minimizing chances of adverse outcomes in the clinic. We are gradually increasing our use of ProteinMPNN and derived ProteinMPNN models in certain fusion protein situations.
A large fraction of biologic therapeutic development is on mAb and IgG fold proteins, which have gone unmentioned in this blog post. Cyrus does not currently work on many mAb or IgG protein folds, although in the past (prior to our pivot away from pure software) we created the nextgen Antibody tool, and we have in the past done some projects using the Rosetta antibody design tool for partners. We anticipate that in the specialized mAb/IgG design space new AI tools will make an increasing impact over time, but have not analyzed those in depth here for this blog post, since these tools don’t match our current commercial priorities.
If you have questions or comments about this post, or would like to discuss a collaboration with Cyrus, please reach out to info@cyrusbio.com. Cyrus pursues internal programs in biologics discovery (current programs are in gout, Fabry disease, gene therapy technology, COVID, and other indications) as well as milestone and royalty based collaborations with other firms (e.g. Selecta), and we are open to new collaborations in indications where our capabilities can help create unique value.
COI Disclosure: Cyrus Biotechnology develops its own Rosetta, Rosetta/AI, and pure AI algorithms for protein design and structure prediction, and has a financial interest in those algorithms and in the biologic assets developed with those algorithms. Cyrus is a co-founder and Executive Committee member of the OpenFold consortium openfold.io, which is part of the 501c3 OMSF, releasing all code under permissive Apache licenses, therefore neither Cyrus nor other OF members have a direct financial interest in OF software.
https://cyrusbio.com/wp-content/uploads/CyrusLogoStaggeredWhite.svg00Thomas Tuonghttps://cyrusbio.com/wp-content/uploads/CyrusLogoStaggeredWhite.svgThomas Tuong2023-03-29 14:35:382023-06-27 11:12:40The impact of new protein AI tools on real-world biologics discovery
Recent advances in the architecture and scale of AI are leading us from the era of narrowly focused AI (e.g. text auto-complete, immune epitope prediction in a protein sequence, antibody domain detection) to broader models with applications across domains from sales and marketing to medical diagnostics. In protein biochemistry these are the “AIFold” models such as AlphaFold2, RoseTTAFold, and OpenFold, which outperform previous models and physics-based systems at structure prediction.
Scientists at Stanford formalized the concept of foundation models in 2021, describing them as AI models “that are trained on broad data at scale and are adaptable to a wide range of downstream tasks”, much as GPT-3 or BERT work as language models. A key aspect of foundation models is that they can be used without further modification or training to perform novel business functions, but they can also be fine-tuned with additional training data to gain greater performance for a highly specialized task – GPT-3 can be provided samples of an individual’s writing to make an AI model whose writing closely resembles that individual. At Cyrus, we see the AIFolds, together with protein design AI and physics-based traditional models, as the foundation models of protein engineering and design, leading to a next generation of new biologics derived either from natural proteins or fully designed from scratch.
In other areas of the software world, foundation models are rapidly becoming cores around which value can be created in individual sectors, such as search, writing assistance, marketing copy, and sales coaching, as described in Forbes. The term foundation model describes both the opportunities and the perils (such as failure in critical use cases or social bias) of the widespread use of tools such as GPT-3. These models are possible because of increases in the scale and speed of GPU (graphics processing units) chips, and innovations in the practice of AI, chief among those the Transformer model. Transformers solve the problem of context, or “attention” in AI terms. In written language, attention is what allows me to write not just the next word or two in this sentence, but to craft a full sentence, situate it in a paragraph, and plan out the flow of that paragraph in an overall piece of writing. It is a major element in what allows AI to appear intelligent. Without attention AI can complete a short phrase, but with a transformer an AI model can write a convincing paragraph.
Rosetta, developed at David Baker’s lab at UW Seattle, combines statistics and atomic-scale physics to model proteins and other molecules, and has been the leading model for protein engineering at the atomic scale for two decades (Molecular Dynamics has been the other dominant model for protein behavior, but not design). Rosetta is not a trained AI system, but instead combines various human-tuned parameters to achieve the highest performance against many benchmarks. Rosetta also achieved many of the firsts in computational protein design, including the first designed protein, top7, and the first designed protein drug, SKYCOVIone. Many groups have built off of the core of Rosetta to make new functions for certain types of molecules. So in this sense Rosetta has served as a foundation model in a pre-transformer world. Baker’s work was honored with the 2021 Breakthrough Prize in Life Sciences, often considered a precursor to a Nobel prize for important new science.
In late 2019, Google DeepMind took a big step forward with the release of the AlphaFold2 protein structure and sequence model for protein structure prediction, borrowing the Transformer concept that had been applied to machine vision and large scale language models and applying it brilliantly to protein systems. RoseTTAFold, OpenFold and others soon followed with similar or identical architecture and scale, each reaching about 1/1000th the size (number of parameters) of well known Large Language Models (LLMs) such as GPT-3, and BERT. Scientists at Meta AI separately trained pure protein language models, without structural information, to even larger scale, with ESM2 reaching about 1/10th the size of the LLMs. In language and image modeling the performance of deep learning models has tended to increase with increasing number of parameters or number of input data points, and for protein models such as ESM2 it has also been true that new behavior emerges at larger scale.
These AIFold models have exploded in popularity across biology. Now extensions to these models, such as Baek and Baker’s RoseTTAFoldNA for nucleic acids, are focusing on individual types of molecules, such as monoclonal antibodies. The Baker lab has also developed another large-scale or “deep learning” AI system for protein design, ProteinMPNN, which is opening up powerful new approaches to better engineering of novel proteins and for improving existing biologics. Unfortunately, for a variety of technical reasons, although AlphaFold2 was the first of the new AIFolds it is not possible to use it to train derivative models with new data. For this reason, systems such as RoseTTAFold and OpenFold are more accessible alternatives for the academic and drug discovery communities.
Now, through the creation of OpenFold with Mohammed AlQuraishi’s lab, along with Genentech, Amazon, Arzeda, Cyrus, Outpace, and some AI non-profit organizations, the Biopharma and Tech communities are creating a trainable foundation model for proteins with state of the art performance that surpasses AlphaFold2 in both speed and memory usage on commercially available NVIDIA chips. AlphaFold2 represented a huge advance, being honored with the Breakthrough Prize for 2023, but it was trained using a proprietary system and with Google proprietary TPU chips that are not available to others – OpenFold creates a truly extensible foundation model that is tuned for training and inference on commercially available NVIDIA chips.
The OpenFold consortium is already working on an extension to the original model that will incorporate larger sets of protein sequences as input, OpenFold-singlesequence. In the future, the consortium will expand to other types of molecules. But the goal of the group is to enable anyone to create derived models from the OpenFold foundation using their own biological data, sequences, structures or whatever data might be developed in the future.
In many cases there won’t be enough data to train useful models, and as Baker and Baek have pointed out, those cases will benefit from a complementary system such as Rosetta. For example, in order for Rosetta to model small molecules it requires molecular modeling software such as RDkit to calculate the physics of the non-protein atoms.
At Cyrus we are embracing all of these future possibilities. We have been working on hybrid Rosetta/AI systems, such as our T-cell epitope deimmunizer, for years. We developed a system to automatically incorporate small molecular calculations into Rosetta, integrating tools from Openeye, to create a hybrid system capable of modeling proteins with small molecule drugs. Now with our unique Deep Mutational Scan capabilities in mammalian cells, and collaborations with over 100 biotech companies under our belts, we can begin to extend OpenFold and other foundation models such as ProteinMPNN into future applications. By doing so we’ll improve our ability to create new biologics from natural proteins, and bring new therapeutics to patients across a wide range of indications from infectious disease vaccines and therapeutics, to rheumatology, to rare disease, and to autoimmune disorders.
About Cyrus Biotechnology
Cyrus Biotechnology is a pre-clinical-stage biotech company combining computational and experimental protein design and screening to create novel biologics for serious unmet medical needs. Using this approach, Cyrus is developing an early pipeline of innovative programs in multiple indications. The Cyrus platform improves both the efficacy (binding affinity, aggregation propensity, solubility, and stability) and safety (binding specificity and immunogenicity) of natural proteins. Cyrus is also partnering with leading biotech and pharma companies and research institutes to bring collaborative programs forward from discovery to the clinic. Cyrus is based on core software from the lab of David Baker at the University of Washington. Cyrus has worked with over 90 industry partners. We are based in Seattle, WA and financed by leading US and Asian Biotech and Tech investors including Orbimed, Trinity Ventures, Springrock, Agent Capital, iSelect, Yard Ventures, WRF, and Alexandria.
https://cyrusbio.com/wp-content/uploads/CyrusLogoStaggeredWhite.svg00Joe Ylaganhttps://cyrusbio.com/wp-content/uploads/CyrusLogoStaggeredWhite.svgJoe Ylagan2022-10-03 12:53:492023-06-06 22:51:23Foundation Models for Proteins: Cyrus, OpenFold and the future of biologics